A major research goal of the Arnaout Laboratory is to elucidate the structure and function of integrins, cell adhesion receptors that play vital roles in normal physiology and disease and use the derived information in structure-based design of new and safer anti-integrin drugs targeting heart disease, fibrosis, and cancer.
Other research interests include elucidating mechanisms underlying cyst formation in Autosomal Dominant Polycystic Kidney Disease, transcriptional regulation of hematopoiesis, mechanisms of kidney regeneration, and design of microfluidic dialysis devices.
Atomic structure of the integrin aVb3 in complex with macromolecular ligands
Structure-based design of novel anti-integrin therapeutics
EM structure of the leukocyte integrin CD11b/CD18 (aMb2)
Role of adaptor proteins in regulation of leukocyte integrins
Role of integrins in ischemia-reperfusion injury models
Mechanisms of cyst formation in Autosomal Dominant Polycystic Kidney Disease
Mechanisms of renal injury and regeneration
Development of microfluidic devices for dialysis
Transcriptional regulation of hematopoiesis
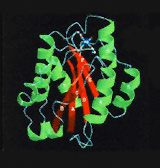
Crystal structure of the A-type domain from integrin CD11b. Cell 1993;72:287, Cell, 1995;80:631
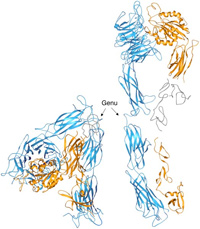
A ribbon drawing of the crystallized extracellular segment of alpha-V beta-3. The alpha-V and beta-3 subunits appear in blue and yellow, respectively. The structure is severely bent at the kneelike "genu" (arrows). Disordered regions are in gray. At right is a computer model of the straightened alpha-V beta-3, which was developed by extension (135 degrees) and rotation (120 degrees) of the bent structure at the genu. The approximate location and shape of three small and disordered domains are shown in gray. Science, 294:339, 2001
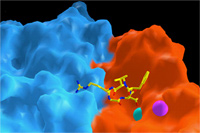
Surface representation of the ligand-binding site in an integrin heterodimer (a subunit is in blue; b subunit is in red) . The ligand is an Arg-Gly-Asp containing peptide, shown as ball-and stick model. Two metal ions are shown in cyan and magenta. Science, 296:151, 2002
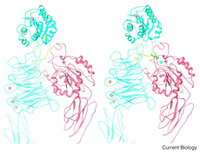
Ribbon diagram of a hypothetical model showing an inactive (left) and an active (right) conformations of the aA-integrin CD11b/CD18. Current Biology, 12:R340, 2002
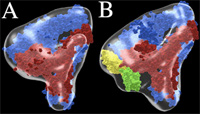
Pseudoatomic models of unliganded (A) and fibronectin-bound (B) integrin aVb3. aV is in blue and b3 in red. Fn-9 and 10 are in green and yellow, respectively. J Cell Biol 168:1109, 2005
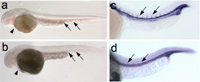
(a, b) Loss of the transcription factor ZBP-89 results in a bloodless phenotype in zebrafish. (a, b) DAF staining of 48 hpf whole-mount zebrafish embryos. Blood (arrows) is present in axial vessels and heart (short arrow) in wild-type (a) but not ZBP-89 morphant (b) embryos. Views are lateral with anterior to the left and dorsal to the top. (c, d) Overexpression of ZBP-89 impairs angiogenesis. In situ hybridization of wild-type embryos (c) or wild-type embryos overexpressing ZBP-89 (d) reveals a marked reduction in intersomitic expression of the endothelial markers flk1 (d), when compared with the respective wild-type embryos. Development, 133:364, 2006
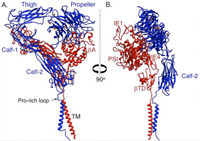
Ribbon diagram showing two views of a structure model of the complete aVb3 ectodomain plus the TM domains. The orientation of the ectodomain relative to the TM domains, show that the ligand binding site is accessible to macromolecular ligands without unbending. The a-genu and propeller metal ions are in orange. J Cell Biol, 186: 589, 2009
Research Positions
If you are interested in applying for a postdoctoral position, or are a Harvard PhD student interested in a laboratory rotation, please e-mail your CV (for student and postdoctoral) and reference letters (for postdoctoral) to: aarnaout1@mgh.harvard.edu
Dr. Arnaout was elected as member in the prestigious American Clinical and Climatological Association (ACCA) in Oct. 2019. Members of the ACCA comprise outstanding physicians selected on the basis of their leadership, scientific excellence, high level of integrity and professionalism. Active membership is limited to 250 physicians.
Dr. Arnaout has received the 2018 Homer W. Smith Award from the American Society of Nephrology (ASN). Read more on the Harvard Medical School website.
Dr. Arnaout has received the 2017 Kuwait Prize in Applied Medical Sciences from the KFAS. Read more in the MGH Awards and Honors newsletter.
Researchers used computer modeling to evaluate the potential clinical impact and cost-effectiveness of administering long-acting, injectable antibodies to infants from birth to prevent HIV infection during breastfeeding.
Uncontrolled blood pressure puts people at increased risk of developing heart disease, brain disease, and kidney disease, yet only one in four people have their blood pressure under good control.
Shadmehr (Shawn) Demehri, MD, PhD, is the corresponding author of a paper published in Cancer Cell, “Commensal papillomavirus immunity preserves the homeostasis of highly mutated normal skin.”
1of
6
About the Nephrology Division
The Division of Nephrology at Massachusetts General Hospital is a leading provider of services for patients with kidney disease, including diagnosis and management of kidney diseases and medical management of renal transplantation.