How TSC Affects Brain Anatomy
Brain involvement is very common in people with TSC and is often the disorder's most pressing concern, having been linked to seizures, cognitive impairment, behavioral disorders, and other neurological complications. Fortunately, despite the fact that approximately 80 percent and possibly a higher percentage of people with TSC have brain involvement, it does not always have debilitating effects.

The brain is one of the most complex organs in the body and is the nervous system's control center. Normally the brain functions as a unified whole, with certain regions specializing in particular functions. However, when this complex structure is altered, the brain may fail to function normally.
There are three main anatomical features associated with TSC that alter the structure of the brain: cortical tubers, subependymal nodules (SENs), and subependymal giant cell astrocytomas (SEGAs). Cortical tubers form in and around the cerebral cortex, the brain's outermost layer. SENs and SEGAs form deeper within the brain, typically along the ependymal lining (walls) of the ventricles, the cavities containing cerebrospinal fluid.
Like TSC lesions that affect other parts of the body, brain lesions associated with the disorder are composed of masses of abnormally shaped, dysfunctional cells. Tubers are composed of cells that fail to differentiate into functional neurons and glial cells during early stages of brain development. The resulting cell masses form before birth and are not thought to increase in size or number over time. In contrast, SENs and SEGAs are benign tumors composed of abnormal cells called neuroastrocytes, which remain in an interior region of the brain called the germinal layer, under the ependymal lining. As tumors, SENs and SEGAs can grow in both size and number over time.
Diagnosis
Tubers, SENs, and SEGAs often play a key role in the diagnosis of TSC. Brain images such as those produced by computed tomography (CT) scans and magnetic resonance imaging (MRI) enable neurologists to identify these lesions and confirm the diagnosis of TSC. All three types of lesions are considered major features in the diagnostic criteria of TSC.
Three Types of Brain Manifestations
Cortical Tubers
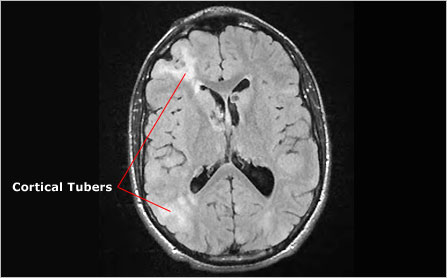
Cortical tubers, from which tuberous sclerosis complex derives its name, are found in more than 80 percent of people with TSC. These benign lesions are found most often in the brain's outermost layer, the cerebral cortex. However, they can also be found in other regions of the brain and in other parts of the central nervous system, including the cerebellum and, rarely, the brain stem and spinal cord. Cortical tubers range in size from a few millimeters to several centimeters in diameter, and people with TSC may have anywhere from 0 to more than 20. Cortical tubers arise during early brain development and are present from birth. They are not thought to change in size or number over time.
Cortical tubers are characterized by the undifferentiated and dysfunctional cells that comprise them. These cells, which have characteristics of both neurons and glial cells, form dense masses that disrupt the highly organized interconnected layers of the cerebral cortex. More importantly, it is thought that they disrupt the functional connections between various parts of the brain, contributing to neurological problems such as epileptic seizures, and learning and behavioral issues. How they might do this is not entirely clear. However, studies have shown a positive correlation between the number of tubers—or, more recently, the volume of brain space occupied by tubers—and the frequency of these neurological problems. The location of tubers may also play a role.
Subependymal Nodules (SENs)
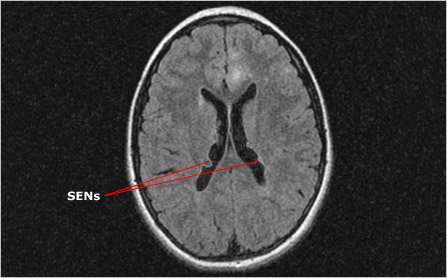
Like cortical tubers, subependymal nodules (SENs) are seen in approximately 80 percent of people with TSC. Typically these benign tumors arise along the ependymal lining (walls) of the lateral ventricles, the spaces that contain cerebrospinal fluid. They vary greatly in size and number, ranging from 2-10 mm in diameter and usually numbering more than one.
And like cortical tubers, SENs form early in brain development and are made up of highly disorganized and dysfunctional cells. However, while tubers have cells with both neuronal and glial characteristics, SENs are composed only of glial cells. SENs also differ from the relatively static tubers in that their growth can outpace that of the surrounding tissue, causing them to protrude into the cavities of the ventricles. While only 15 percent of SENs grow larger than 1 cm in diameter, those that do become classified as SEGAs and are cause for concern.
Subependymal Giant Cell Astrocytoma (SEGA)
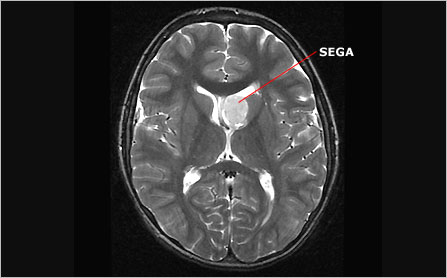
Subependymal giant cell astrocytomas (SEGAs) are large SENs. They too are benign tumors composed of undifferentiated, dysfunctional glial cells. However, because of their large size and their potential for continued growth, especially in children and adolescents, these lesions pose a significant risk. For reasons that are not well understood, the propensity for SEGAs to develop decreases dramatically after adolescence.
In rare cases, SEGAs grow large enough to obstruct the flow of cerebrospinal fluid through the lateral ventricles. This is more likely when they occur in particularly narrow passages such as the foramen of Monro, which carries fluid between the lateral ventricles and the third ventricle. Obstruction of this flow can cause a condition known as hydrocephalus, in which cerebrospinal fluid builds up causing the ventricles to expand, and pressure within the cranium to increase. Without intervention to relieve the pressure, hydrocephalus can cause permanent damage to the brain or, in rare cases, death.
Follow-up and Treatment
Although brain lesions are common in people with TSC, their effects vary greatly. Some people with TSC-related brain abnormalities experience few or no debilitating neurological complications for the duration of their lives. However, because of the possible connection between cortical tubers and epileptic seizures, and because SEGAs are potentially life threatening, it is important for people with TSC to undergo regular brain imaging and examinations by a neurologist who specializes in the disorder.
Upon diagnosis, physicians and imaging specialists use computed tomography (CT) scans or magnetic resonance imaging (MRI) to identify any and all brain lesions. This initial examination establishes a baseline against which all future examinations can be compared. In most cases, brain imaging for TSC should be repeated every one to three years through childhood and adolescence. Specialists recommend more frequent examinations for individuals with SEGAs. If a SEGA is present, examinations as frequently as every three to six months may be necessary to carefully monitor the tumor for further growth and/or obstruction. This is especially true during childhood and adolescence, when SEGAs are most likely to grow. For reasons that remain unclear, SEGAs lose their propensity for growth during late adolescence.
Doctors may also use an electroencephalogram (EEG) examination to assess the electrical activity in the brain if there is a concern that an individual is experiencing seizures. Individuals with TSC should be aware of the ongoing risk of seizures and discuss with their physician any concerning sensations or behaviors they may have.
Surgery
Surgical intervention for brain abnormalities is usually not necessary. However, large, progressive SEGAs that obstruct the flow of cerebrospinal fluid and increase intracranial pressure present a neurosurgical emergency and must be removed. Surgery typically provides a permanent solution to this serious medical condition—but not always. As told in Michael's family story, SEGAs can regrow following successful surgery and sometimes need to be removed again. Also, in cases involving SEGAs that are particularly large or otherwise difficult to remove, the flow of cerebrospinal fluid may remain obstructed following surgery. In such cases, neurosurgeons place a section of tubing, called a shunt, into the obstructed section, so that fluid can flow freely and pressure does not build up.
People who suffer from intractable seizures may also be treated surgically, provided the source of the seizures is localized to a specific region of the cerebral cortex, usually a cortical tuber. Neurosurgeons have successfully reduced or eliminated seizures in some people by removing such tubers. However, it is not always possible to determine which tuber or part of the brain might be responsible for seizures.
Next Steps
It is important to remember:
- There are three types of benign brain findings associated with TSC: cortical tubers, subependymal nodules (SENs), and subependymal giant cell astrocytomas (SEGAs)
- Cortical tubers are typically found in the cerebral cortex, the brain's outermost layer, and may be associated with seizures, learning difficulties, and behavioral problems. They are present at birth and are not thought to grow
- SENs and SEGAs are typically found along the walls (subependymal lining) of cavities (ventricles) deep inside the brain. If they grow large enough, they can obstruct the flow of cerebrospinal fluid through these cavities
- TSC specialists recommend brain imaging every one to three years during childhood and adolescence, and more often for people with progressive SEGAs. SEGAs lose their propensity for growth during late adolescence for reasons that remain unclear
- It is important to find a neurologist who is familiar with TSC-related brain abnormalities and their neurological consequences