
Krantz Family Center for Cancer Research
Haas Lab


Haas Lab
Explore the Haas Lab
2024 Krantz Awards Recipient
2024 Breakthrough Award: Discovering how functional molecular networks are driving treatment resistance in certain cancers
Team: Wilhelm Haas, PhD, Othon Iliopoulos, MD, Mo Motamedi, PhD.
Learn about the team's project and the Krantz Awards
Research Summary
Proteins are the central molecular players in virtually all biological processes and cellular functions. The proteome is the entirety of all proteins in a biological system. The proteome’s vast complexity is not only defined by the number of expressed genes but also driven through posttranslational protein modifications - such as phosphorylation – and the interaction of proteins to form functional units in the form of multiprotein complexes. The Haas laboratory utilizes mass spectrometry-based proteomics to decipher this complexity. The lab aims to understand the extensive changes the proteome undergoes in cancer and to leverage these changes for early cancer detection, cancer diagnosis, cancer treatment guidance, and the development of new treatment strategies.
Research Projects
Cancer is based on dynamic changes of the genome that ultimately translate into an altered proteome, optimized for uncontrolled cell growth and division. In addition, many pathways, initially causing cancer, further promote the propagation of altered genetic information, accelerating the adaption of cancer cells to new environments. This dynamic process becomes even more complex if taking into account the dynamic state of the cellular proteome that is regulated by protein synthesis and degradation, post-translational modifications, protein localization, and the interaction of proteins with other proteins, as well as with different classes of biomolecules. While the cancer genome is now established as a source for cancer diagnosis and for directing treatment strategies, we are only beginning to tap into the information contained in the cancer proteome. Yet, the proteome holds enormous potential to improve our understanding of the basic principles underlying cancer, to revolutionize the early diagnosis of the disease, and to improve patient care. To date, virtually all targeted therapeutics in cancer treatment are targeting proteins. Understanding how these drugs alter the proteome and the interactome – the global map of protein-protein interactions – has the potential to help us refine our approaches to drug design.
The core technology used in our research group is high-throughput quantitative proteomics enabled through multiplexed mass spectrometry. Sample throughput is a key requirement in cancer proteomics as it allows handling the analysis of the large number of samples that have to be examined to generate the basis for understanding a disease that displays such heterogeneity. Foremost, throughput is essential in the early detection of cancer through mapping blood plasma proteomes to detect cancer biomarkers. If such assays are successful, they will eventually be used to map millions of blood plasma samples. To enable such applications of mass spectrometry, we have developed a novel high-throughput proteomics platform including an autonomous artificial intelligence (AI)-powered mass spectrometry data acquisition method to enable unbiased deep proteome mapping. Unbiased screening of >2000 proteins from blood plasma samples (in 10 minutes per sample) rather than mapping a small number of biomarkers will allow us to enable a multi-biomarker assay for multiple cancer types that is constantly improved through adaptation to the detection accuracy.
This technology also allows mapping >8000 proteins of cancer cell line or tumor tissue. samples at the same high throughput. Analyzing the proteome maps across a panel of cancer cell lines, we recently observed that the concentration of proteins in known complexes is accurately correlated across all analyzed cell lines. We showed that protein co-regulation analysis allows the genome-wide mapping of proteinprotein interactions with an accuracy ten -times larger than that when using coexpression analysis based on RNAseq data. We further found that deviations from co-regulation of two interacting proteins in specific cancer cell lines reflect perturbed cellular circuitry, and it remarkably predicts sensitization to therapeutics targeting regulatory modules in the associated pathway. We have termed this approach to fast, in-depth characterization of protein-protein interaction landscapes interactome dysregulation (DysReg) mapping. This novel method enables an interactome-wide mapping of protein-protein interaction dysregulation and inferred cancer vulnerabilities of any cancer sample based on a proteome map acquired at high throughput.
Our goals are to apply these technologies to (i) identify novel cancer vulnerabilities that direct new treatment strategies, to (ii) map cancer vulnerability dynamics, such as those occurring in the development of therapy resistance, to identify novel targets that enable to overcome the treatment resistance, and to (iii) use our technology in a clinical setting for mapping tumor vulnerabilities to inform treatment strategies in a patient-specific manner.
We also recently identified the E3 ligase UBR4 as a key regulator in adjusting the concentration level of interacting proteins – the molecular mechanism enabling our interactome mapping – and we have shown that this role presents UBR4 as a target for treating aneuploid cancer.
Instruments
 The Haas laboratory's mass spectrometers include a Thermo Scientific Orbitrap Astral, acquired through NIH Shared Instrumentation Grant (S10) funding. The Astral enables high-resolution, high-sensitivity proteomic analysis at unprecedented acquisition speeds, enabling deep, quantitative proteome coverage of complex biological samples. The instrument supports researchers pursuing large-scale proteomics, single-cell analysis, and other applications requiring rapid, high-throughput mass spectrometry. Please send project inquiries to whaas@mgh.harvard.edu.
The Haas laboratory's mass spectrometers include a Thermo Scientific Orbitrap Astral, acquired through NIH Shared Instrumentation Grant (S10) funding. The Astral enables high-resolution, high-sensitivity proteomic analysis at unprecedented acquisition speeds, enabling deep, quantitative proteome coverage of complex biological samples. The instrument supports researchers pursuing large-scale proteomics, single-cell analysis, and other applications requiring rapid, high-throughput mass spectrometry. Please send project inquiries to whaas@mgh.harvard.edu.
Research Positions
Postdoctoral fellowships in cancer proteomics
The Haas Laboratory at the Mass General Brigham Cancer Institute and Harvard Medical School, Boston, MA, United States, is seeking highly-motivated candidates for two postdoctoral positions. We are leading experts in the field of quantitative mass spectrometry-based proteomics, and we are seeking candidates for two distinct projects.
Project 1: Identify cancer vulnerabilities suitable for personalized cancer treatment strategies. We have developed a method to use quantitative proteomics for high-throughput mapping of global protein-protein interactions landscapes (interactomes) (Lapek et al. (2017) Nat. Biotechnol. 35(10):983-989). Dysregulation of the interactome is a hallmark of cancer, but the specific functional consequences are poorly understood. This project aims to map dysregulations of specific protein-protein interactions in cancer to identify those that can be exploited for treatment. Candidates with a PhD or MD/PhD in biology, biochemistry, chemistry, oncology, immunology, or a related field are encouraged to apply. Strong technical expertise in biochemistry and molecular biology is required. Experience in working with mouse models is desired. Candidates with experience in mass spectrometry-based proteomics are preferred but the experience is not required. Candidates should show a strong interest in learning new techniques and an interest in working with big datasets.
Project 2: Early cancer detection through plasma proteomics. High-throughput proteomics has the potential to revolutionize early detection of cancer. We seek to develop new methods in high-throughput mass spectrometry-based proteomics for the mapping of blood plasma samples with the goal of identifying biomarkers for early detection of cancer. The optimal candidate has a PhD in chemistry, physics, engineering, machine learning, computational sciences, or a related field. Candidates should have a strong background in programming. Experience in working with mass spectrometers is desired. Candidates should show a strong interest in working with large datasets and on biological and clinical questions.
We are a multi-disciplinary team across biology, chemistry, and bioinformatics, and the lab is equipped with multiple state-of-the-art, high-performance mass spectrometers. The laboratory is supported by several funding sources from NIH and industry.
Interested candidates should send a cover letter, their CV, and contact information of at least two references to Wilhelm Haas (HaasPostdocSearch@mgh.harvard.edu).
Publications
Selected Publications
Kathiresan M, Animesh S, Morris R, Kreuzer J, Patra KC, Shi L, Merritt J, Yin X, Benes CH, Bardeesy N, Haas W. Protein interactome homeostasis through an N-recognin E3 ligase is a vulnerability in aneuploid cancer. bioRxiv. 2023 May 4: 2023.05.04.539299.
Kreuzer J, Edwards A, Haas W. Multiplexed quantitative phosphoproteomics of cell line and tissue samples. Methods Enzymol. 2019; 626, 41-65.
Lapek JD, Greninger P, Morris R, Amzallag A, Pruteanu-Malinici I, Benes CH*, Haas W*. (2017) Detection of dysregulated protein association networks by high-throughput proteomics predicts cancer vulnerabilities. Nat. Biotechnol. (in press)
Edwards A, Haas W. (2016) Multiplexed Quantitative Proteomics for High-Throughput Comprehensive Proteome Comparisons of Human Cell Lines. Methods Mol. Biol. 1394,1-13.
Braun, C.R.*, Bird, G.H., Wühr, M., Erickson, B.K., Rad, R., Walensky, L.D., Gygi, S.P.*, Haas, W.* (2015) Generation of Multiple Reporter Ions from a Single Isobaric Reagent Increases Multiplexing Capacity for Quantitative Proteomics. Anal. Chem. 87, 9855-9863.
McAlister GC, Nusinow DP, Jedrychowski MP, Wühr M, Huttlin EL, Erickson BK, Rad R, Haas W, Gygi SP. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem. 2014; 86, 7150-7158.
Ting L, Rad R, Gygi SP*, Haas W*. (2011) MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 8, 937-940.
*Co-corresponding authors
Research Image
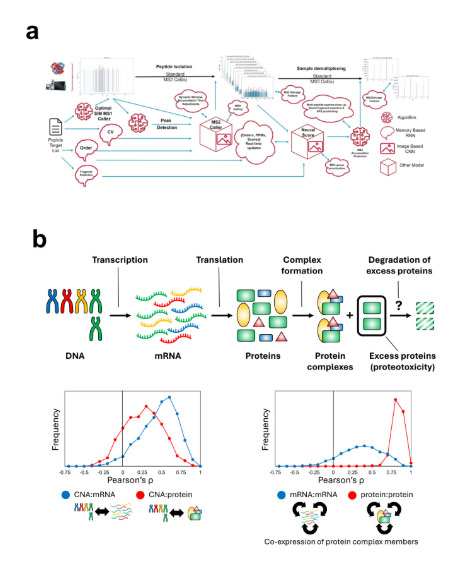
(a) This network of multiple neural networks and algorithms enables ultra-high throughput deep mass spectrometry-based proteomics through AI-directed autonomous data acquisition. (b) In aneuploid cancer, mRNA levels accurately reflect the underlying gene copy alterations, while protein levels are adjusted to the concentrations of the protein interaction partners. This enables the use of protein concentration co-regulation analysis for highthroughput exploration of cancer interactomes. The molecular mechanism underlying the protein interaction-driven concentration regulation reveals potential new treatment strategies for aneuploid cancers.
Our Researchers
Wilhelm Haas, PhD
Group Members
- Sambhavi Animesh, PhD
- Adrian Braun
- Soroush Hajizadeh, MSc
- Annika Hanraths, BSc
- Pranjal Umesh Kalekar, BTech
- Johannes Kreuzer, PhD
- Robert Morris, PhD
- Eric Zaniewski, BSc